Note
Go to the end to download the full example code
Transglutaminase 2: Flexible fitting with AV restraints¶
transglutaminase 2 (TG2)
IMP.bff includes the command line tool imp_bff that
contains the program flexfit - a software to sample and
optimize biomolecular structures with flexible regions based on
experimental information such as fluorophore distances in a protein.
The output of imp_bff flexfit can be visualized in Chimera.
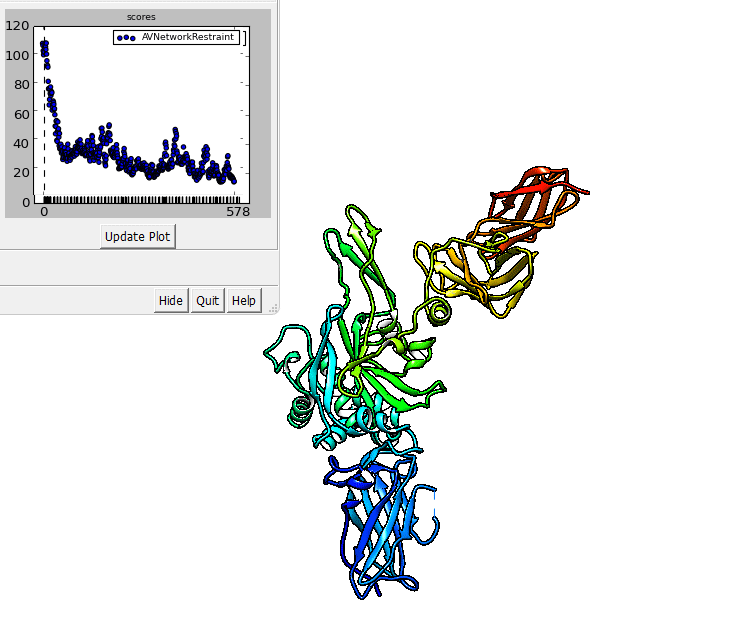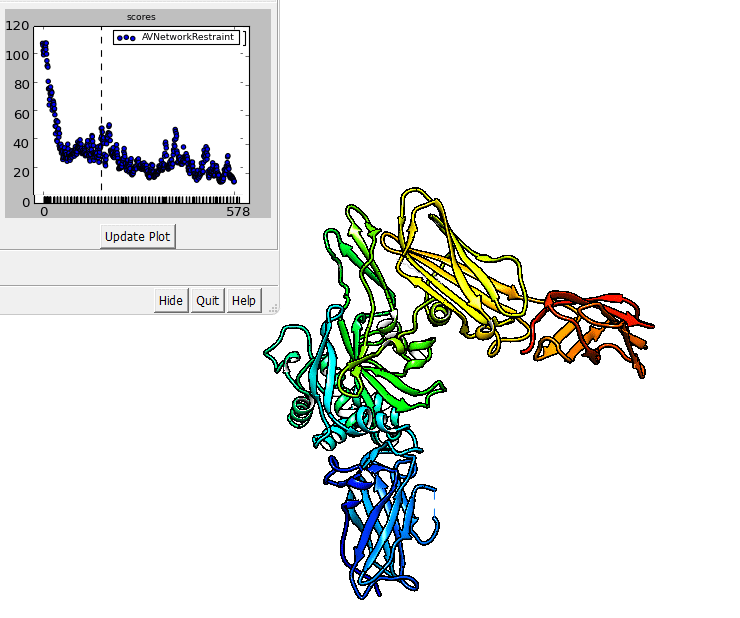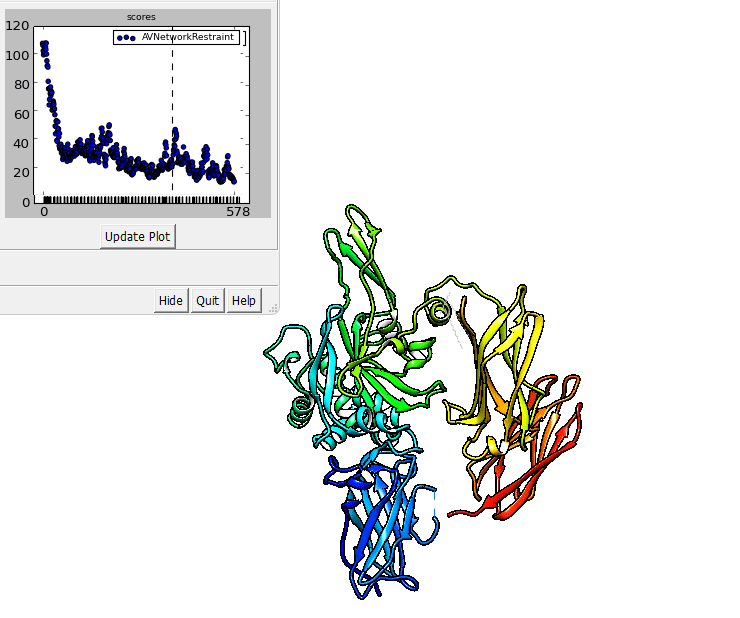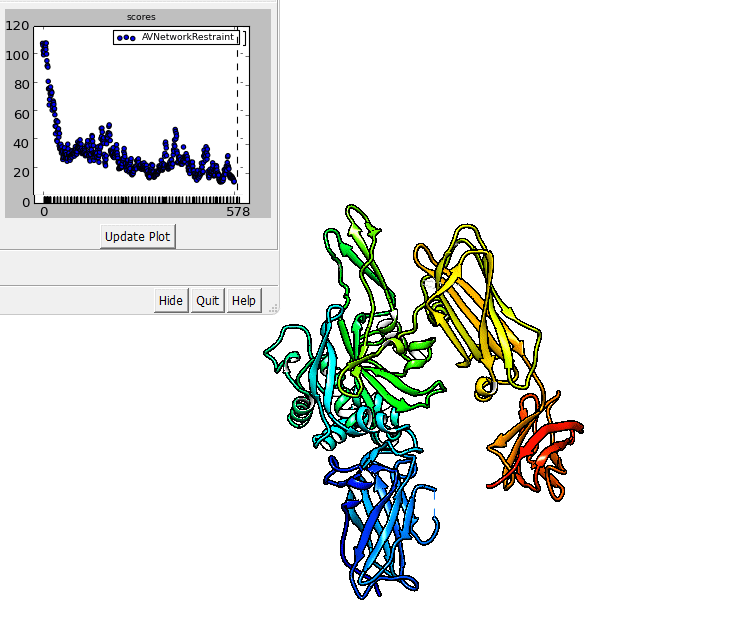
imp_bff flexfit uses an initial structure, a flex.fps.json file. A
flex.fps.json file contains information on inter-label distances and flexible
regions.
For full help on the rrt_sample tool, run from a command line:
$ imp_bff flexfit --help
Setup¶
First, obtain the input files used in this example and put them in the current directory, by typing:
$ cp "<imp_example_path>"/bff/structure/TG2/*.* .
(On a Windows machine, use copy rather than cp.) Here, <imp_example_path> is the directory containing the IMP example files. The full path to the files can be determined by running in a Python interpreter.
Calculation¶
Structure of the protein (transglutaminase 2) that are consistent with
additional experimental information can be sampled running imp_bff flexfit.
First, we need to gather experimental information (here inter fluorophore distances)
and define flexible regions. Labeling sites, distances between labeling sites, and
flexible regions are listed in the input file flex.fps.json.
A flex.fps.json file can contain multiple sets of distances. Thus, distance set used
for scoring must be defined via the -c option. Moreover, a flex.fps.json
file can contain multiple sets of flexible residues (this requires to define the used
set of flexible resiudes via the -f option.
$ imp_bff flexfit -i topology.pdb -l flex.fps.json -c S1_chi2 -f FexResSet1
imp_bff flexfit can operate in a slow and in a fast mode. In the slow mode the
accessible volume (AV) of the labels is computed at each iteration. In the fast mode, the
AVs of the labels are computed once, the mean label position becomes part of a rigid
body and the distance between the mean positions is used for scoring (using a transfer function
that transforms distances between mean positions into experimental observables).
The output files are RMS files that can be inspected in Chimera.
Total running time of the script: (0 minutes 0.000 seconds)